

It's been a year since the FDA first announced its intention to harmonise its 21 CFR 820 requirements with the international medical device standard, ISO 13485:2016.
Standardisation is only a good thing in the long run for medical device companies aiming for GMP-compliant quality management systems.
In the short term, though, the changes spell uncertainty for what good practice in the medical device space actually means.
The full scope and direction of the FDA's transition isn't yet fully defined, leaving some space for guesswork about how exactly CFR 820 compliance is going to change.
But by unpicking the main differences between 21 CFR 820 and ISO 13485 as they stand, we can anticipate some of the key changes.
Qualsys Head of Quality Assurance Kate Armitage runs through the three main differences we can expect in the new medical device world order.

1. More focus on risk
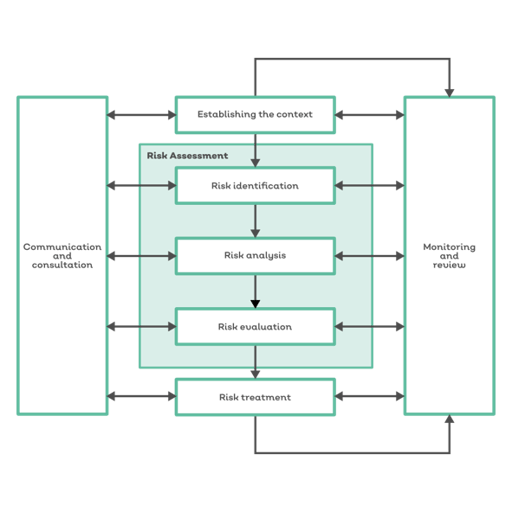
ISO 13485:2016 is built around a risk-based approach for medical device manufacturers.
ISO 13485-compliant manufacturers need to consider how risk is managed for the entire lifecycle of their medical device product.
And risk is central to the ISO 13485 design process in several ways:
- Risk management outputs can be recorded as valid design control inputs
- Elements for safe operation must be identified and integrated into the design process
- Safety requirements and associated risks must be documented as part of the design procedure

FDA regulations currently do not involve such an emphasis on a risk-based approach, only briefly mentioning risk analysis in relation to the design control process and not officially codifying any risk management activity.
With risk-based thinking becoming increasingly part and parcel for governance and compliance, we can expect risk to be central to the new revision of CFR Part 820.
In particular, we can expect ISO 13485's emphasis on iterative risk management, where risk controls are fed back into design controls in a Plan Do Check Act cycle, to become key.
 Need some help with risk?
Need some help with risk?
Watch our ISO 31000 risk management webinar recording
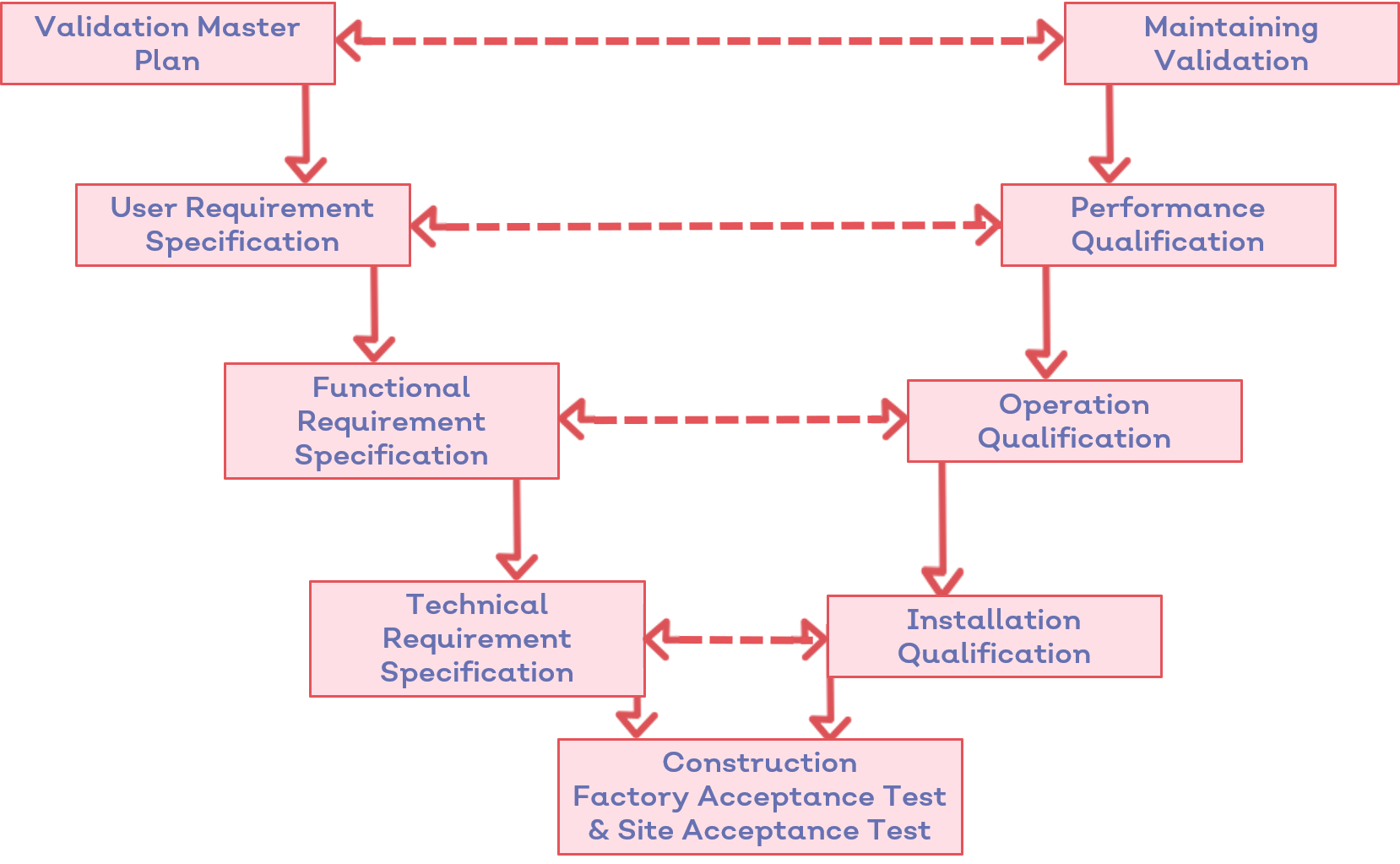
2. More traceability
The FDA does not mandate that you be able to trace your medical device design requirements between stages.
Nor do you have to be able to trace design inputs to outputs in your device history file or 510(k) submission.
ISO 13485, on the other hand, pinpoints traceability as a key requirement for the entire lifecycle of the medical device product.
Those already working to ISO 13485 will be familiar with the process, and will have a documented, traceable journey from design input to output in their design history file.
The FDA's design controls regulation recommends this level of traceability, but doesn't currently make it mandatory.
But with the FDA looking to borrow from ISO 13485, the emphasis on traceability is a key ingredient which could form part of the transition.

Learn more about traceability, design controls,
510(k) and more in our York Instruments case study video
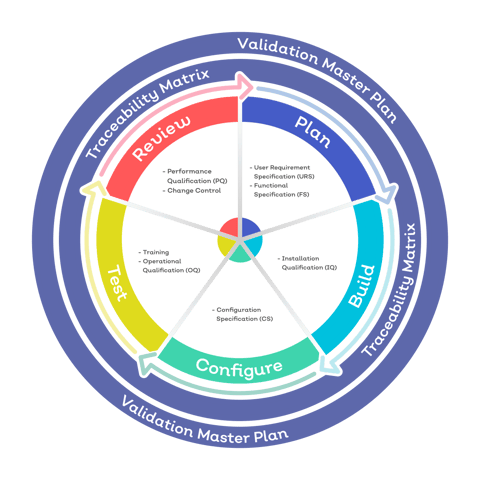
3. More stringent requirements for software validation
Medical device design and manufacture is among the most complex and highly regulated of business activities.
So it isn't surprising that manufacturers are adopting computer and software systems to automate and control the bulk of the quality management process electronically.
The FDA's requirements scope for how those systems are validated for operation in the industry is currently significantly narrower than that of ISO 13485.
The FDA's current focus is on how electronic records are controlled within a design controls software package, and validation centres around the intended use of that software.
Harmonising with ISO 13485 and the risk-based methodology we've already seen would mean a shift in how FDA software validation takes place.
Risk would move to the forefront of the validation process and medical device companies would have to coordinate with their software vendor to unpick the risks, and corresponding risk controls, associated with their software project.
Any QMS software, not just that related to design control management, would fall into this new and broader validation scope.
Learn more about life science software validation
Next steps
Dive deeper into the requirements of ISO 13485 with our free toolkit:




Share your thoughts on this article