

To stay competitive, reduce overheads and scale the business, most manufacturers rely on an advanced and integrated portfolio of automation tools.
When the manufactured products have a clinical, pharmaceutical or medical device use, the manufacturer must be able to demonstrate that the software is fit for purpose and will not impact patient safety.
The role of the quality manager is core here. But it's also very complex when it comes to implementing and managing new software tools for pharmaceutical environments.
You need to know what the tool will do, what processes it will impact and how you're going to bring your people together to effectively demonstrate and document the software is fit for purpose.
The below article is a transcript from Qualsys' recent Quality in Life Science event. In the presentation Hilary Mills-Baker (Emerson) and Karen Ashworth (KACL), go 'back-to-basics' - discussing everything from what regulators are looking for when you implement systems to useful guidance articles, tips to identifying the critical functions to how you can demonstrate your controlling the risks.
|
Want all the resources from the GxP Life Science event? Complete this form:
|
Contents:
- An overview of GAMP
- What is data integrity
- What a manufacturing system does
- Identifying the critical functions
- Managing the life cycle of the system / software that controls the functions
- The risks associated with the functions and how are they controlled
Below is a transcript from the presentation:
Overview of GAMP
GAMP stands for Good Automated Manufacturing Process. The GAMP 5 A Risk-Based Approach to Compliance GxP Computerised Systems was first introduced in 2008. It provides guidelines for life science organisations so they can use automated systems in a compliant manner.
You'll notice in the secondary title it says a risk-based approach. The risks we're talking about are patient safety, product quality and data integrity. These risks come up 54 times in GAMP 5 - I counted them! So these guidelines are all about ensuring automated system risks are controlled throughout the life science manufacturing process.
GAMP 5 guidelines were there from 2008. Since this time, the regulator started to think that although businesses were very focused on patient safety and product quality, data integrity was overlooked and a bit misunderstood. So ISPE responded in 2017 with the "Records and Data Integrity Guide". The guidance aims to help organisations understand data integrity and how apply best practices.
In 2019 there was practical guidance on "Data integrity -manufacturing records." This is a sister guide for GAMP 5 to help you to apply the requirements.
Today we are going to focus on data integrity.
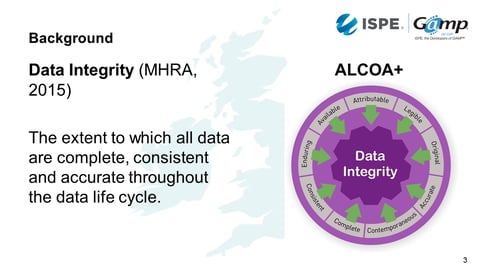
What is ‘data integrity’?
The MHRA defines data integrity as the extent to which all data are complete, consistent and accurate throughout the data life cycle.
From the moment the data is created to twenty years time if a regulator wants to go into the records - it all needs to be managed.
The components of data integrity can be expressed using ALCOA+. This is the acronym for:
- Attributable: Must know who created or modified data
- Legible: Must be readable from creation through to archiving
- Contemporaneous: Dated at the moment of generation / change
- Original: The first recorded value
- Accurate: Its precise and correct
- Complete: Its all there and the units are visible
- Consistent: Made in a repeatable way
- Enduring: Recorded permanently and not lost during the life cycle
- Available: Find the data for an inspection
If we've got all of these, we should have good data integrity.
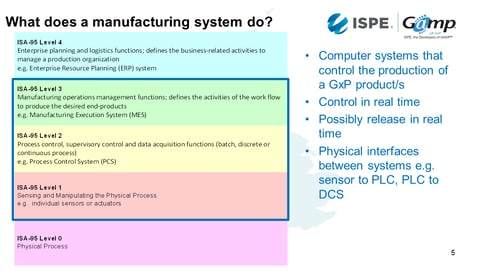
What does a manufacturing system do?

What data does it collect, manipulate or store?
What records are we interested in from our manufacturing systems?
1. Values captured direct from the manufacturing process
The first will be the values captured directly from the manufacturing process such as temperatures, pressures and flow rates. There can be more complex data capture, such as images. There can also be derived process values from multivariate analysis or process modelling.
2. Values received via interfaces from other systems
The second might be data coming from other level systems such as an ERP - such as what batch was made, when and where.
There may also be information from systems which are used in managing materials, equipment and personnel. Are the ingredients in date? Is it within specification? Have we got appropriately trained people available?
There might also be upstream or downstream manufacturing activities. Maybe we have laboratory results that need to come back to us in order to ensure the processes work.
3. Values entered by operations personnel
A typical example is ingredient dispensing details: How much should people be taking, the lot number etc.
These are also records of things that are done manually such as cleaning operations.
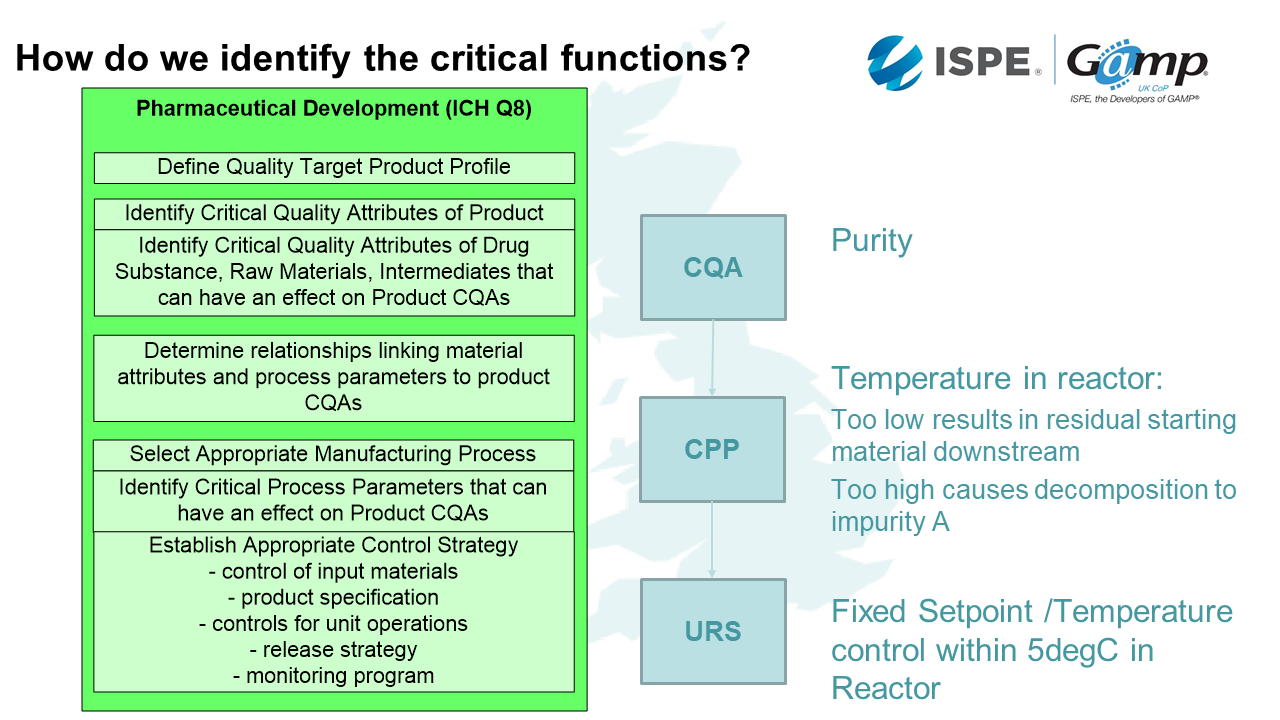
How do we identify the critical functions?
The next thing we need to consider is that is what are the critical functions? We start off looking at our Target Product Profile.
Is it something you rub on, take orally or inject?
That is going to have a lot of associated attributes. How strong will it be and what will the required dosage be? We need to use the product profile to decide what is really critical to product quality.
Then we need to look at the process of how we're going to make this drug. Within that process, there are things which will be critical because they effect the performance.
An example might be a drug whereby the critical quality attribute (CQA) is purity. A critical process parameter (CPP) will be the temperature in the reactor. The purity is controlled by the temperature in the reactor. During the action, if the temperature is too low, this results in residual starting material downstream. If the temperature is too high, it causes decomposition to an impurity. Therefore, if we control this process parameter we can control the purity of that product. There is always a link between the CPA and CPP. How do we then put that into our computerised system? We need a fixed set point and temperature which controls the temperature withing 5degC in the reactor. That's how we can ensure there is purity in my drug.
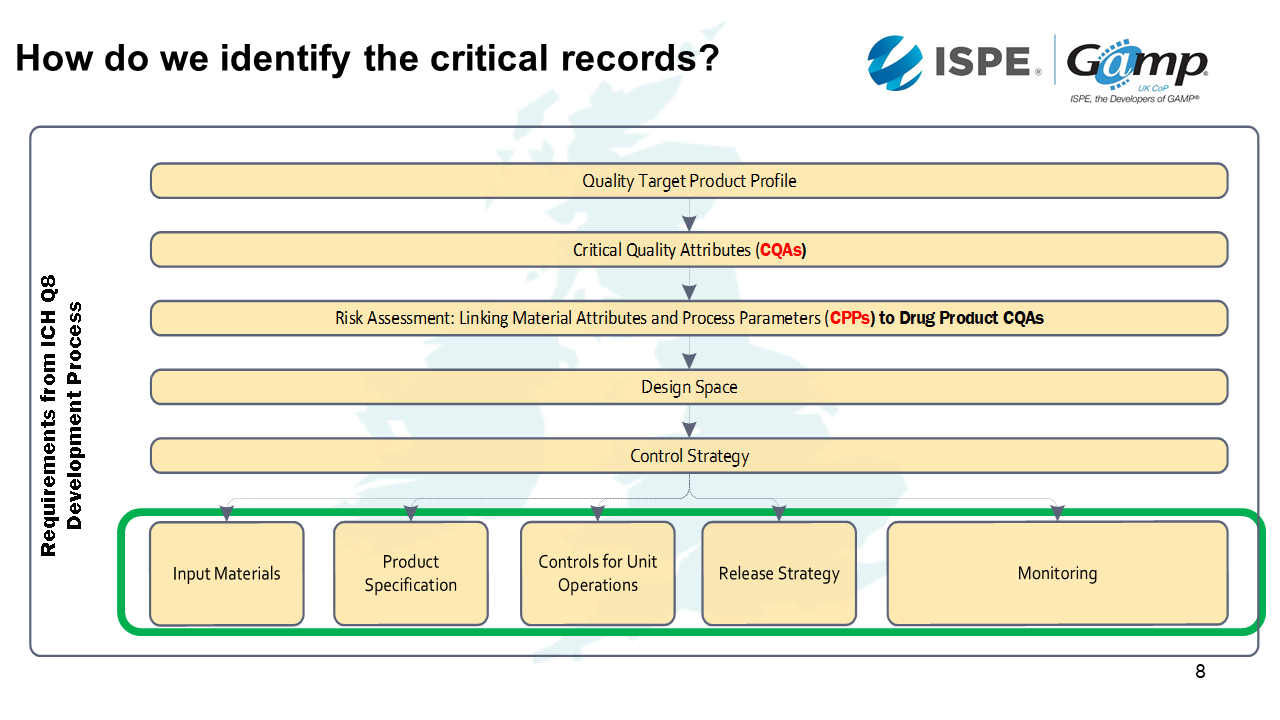
As well as identifying the critical functionality, we need to identify the critical records. These are the things which have an impact on patient safety and product quality. If you've done the ICH Q8 development process properly, the critical records will be evident from that.
The first step in the process is to identify the Quality Target Product Profile. For example, we will need to know whether it's going to be a sterile dosage or through a device. This will then inform the Critical Quality Attributes.
As we start to identify the Critical Quality Attributes, such as the physical, chemical, biological and microbiological things that are important for our product. For example, if we knew it was going to be a sterile dose, we know we are going to need clean rooms and environmental records.
Then when we need to link the quality attribute parameters with the drug product critical quality attributes. If we keep with the clean room example, we're going to have differentiated pressures between the rooms to report.
Then we come into the Design Space. This is when we ask questions such as how much can the parameters move around and what exceptions do we tolerate?
Then we plan the Control Strategy. This is where we look at input materials, product specification, controls for unit operations, release strategy and monitoring.
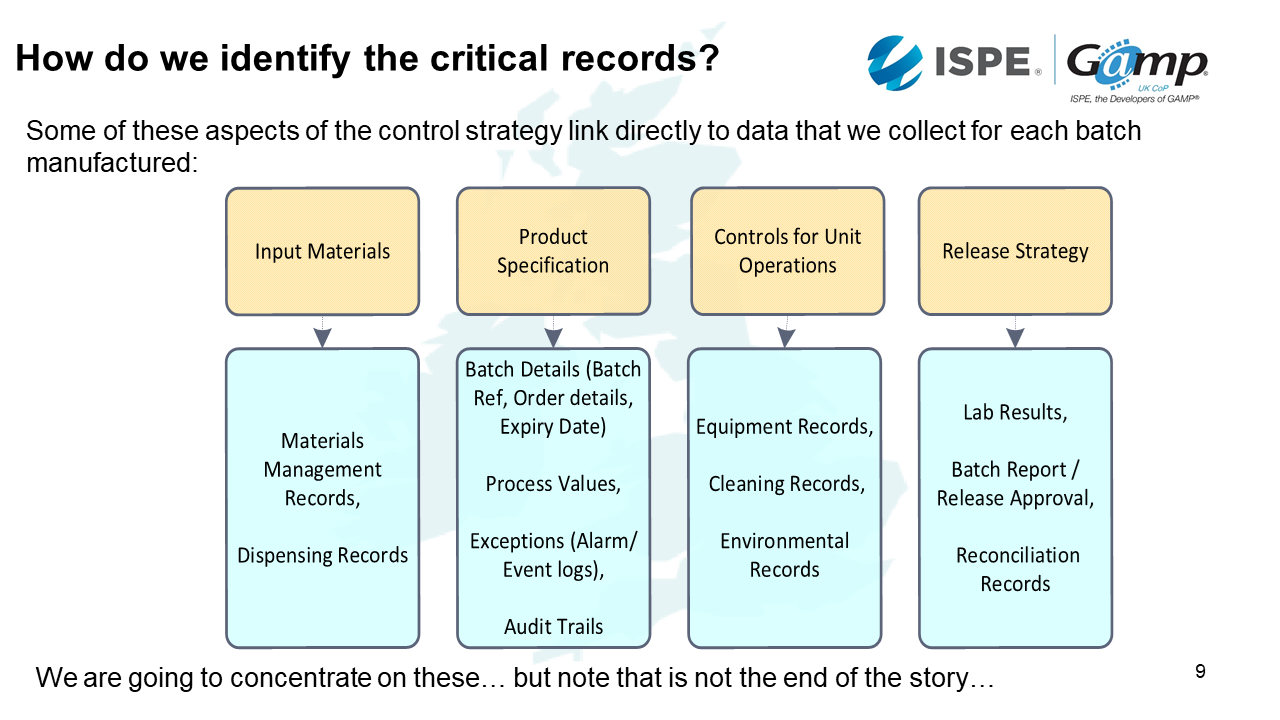
Some of the critical records link to data that we collect for each batch manufactured. Examples are expiry dates and quantities.
For product specification, there are different aspects to this, such as the batch details, process values, exceptions, and audit trails. Then for the release strategy will almost certainly have laboratory results, batch report and reconciliation records.
That's what we collect for a batch, but that is not the end of the story.
Controls for unit operations include things such as equipment records, cleaning records and environmental records.
We collect data for each batch to demonstrate that the necessary controls were effective and the critical process parameters were in specification:
- Input materials: Dispensing the ingredient identity, weight.
- Product specification: Records of processing phases, time sequence, process values, exceptions.
- Controls for Unit operation – records of manual activities like cleaning.
- Release: Records drawn together into a final batch report ; evidence of it having been appropriately reviewed
The ‘per batch’ data is not sufficient in its own right to say that good quality product is being made.
Each aspect of the control strategy also involves some elements that we set up and validate prior to use and/or which we need to monitor to ensure the system and process remain in a validated state:
Input materials:
Master recipe – and route by which control recipes for specific batches are produced from it (interfaces between systems? Automated or manual data transfers?)
Control for Unit Operations – system configurations, SOPs for manual activities
Release strategy – ensuring that all the bits we need to pull together for someone to review do in fact reflect the way the batch was processed
Monitoring - may be independent monitoring of process (those product checks mentioned before for predictive algorithms… or something as simple as CCTV footage of people carrying out processing steps).
Also important to realise that where we are relying on automated systems doing activities we have validated, maintaining those systems in a validated state is critical – hence GAMP O appendices.
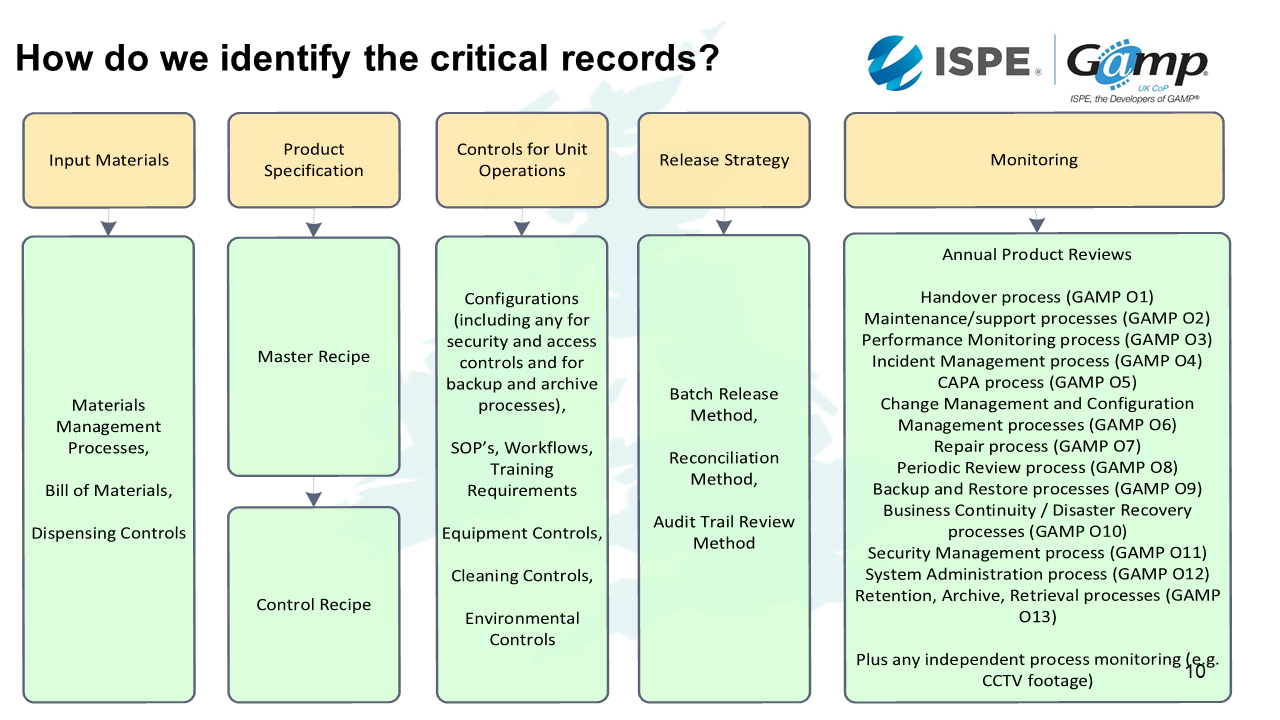
Input materials
In terms of our input materials, these are things we need to set up in advance, check and validate for the batch.
This includes:
- Materials management processes
- Bill of materials
- Dispensing controls
- SOPs
- Calibration
- Barcode scanning of ingredient information
- Traps for out of date or out of specification ingredients
Product specification
Product specification is our master recipe that we need to check it's set up properly and controlled. If we make changes, we need to make sure that they are consistent in subsequent batches. Our control recipe for a batch will be generated from a master recipe.
Monitoring controls
It's really important to have monitoring processes in place. The GAMP operational processes can be very helpful here. For example, I was called out to a system and someone said "my system is really slow at producing strengths. What had actually happened is, people had made minor modifications over several years. They had all been very minor so every member of staff hadn't gone back to check the impact of their modifications on the overall capacity or processing speeds. It eventually got to the stage where the processing speed wasn't fast enough for it to be able to record the check when it needed to. That meant that the index wasn't able to pull all the data as a full string which was why it was slow. In an adequate change and configuration management process, regression analysis would have checked this. An adequate annual performance monitoring process should have also raised this.
Backup and restore and business continuity processes shouldn't just check that you've got the functionality back, you should also have the data available. For example, a PLC was collecting data every month, when the storage got full the IT team collected the data and archived it. They, however, then had a reorganisation. This meant that the process just got forgotten about and the data was just being overwritten month after month. It's really important when managing records that we remember things change and we need to keep control.
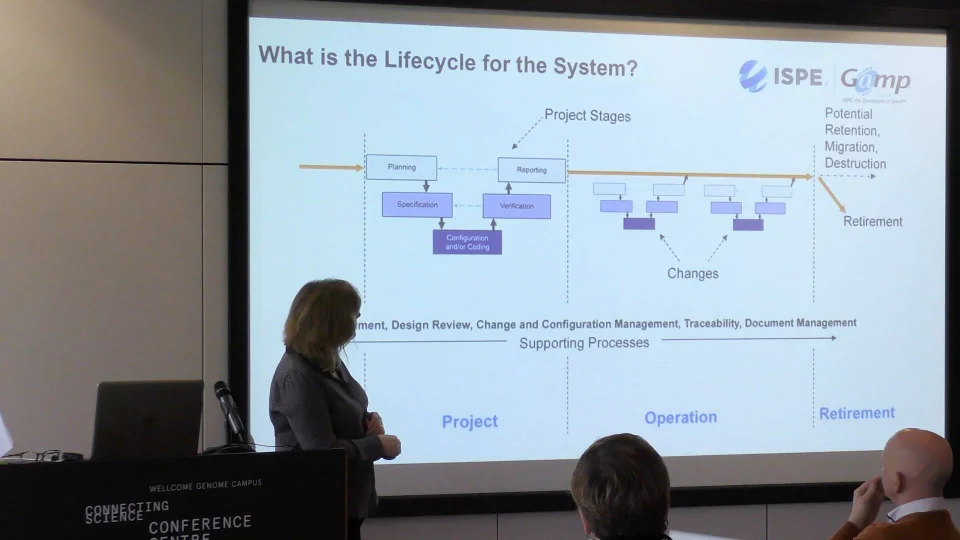
What is the life cycle for the system?
So far we've covered what is critical about the functionality of our computerised system and about the data. Now we are going to look at how we develop our manufacturing computerised system to make our product.
This lifecycle is from GAMP:
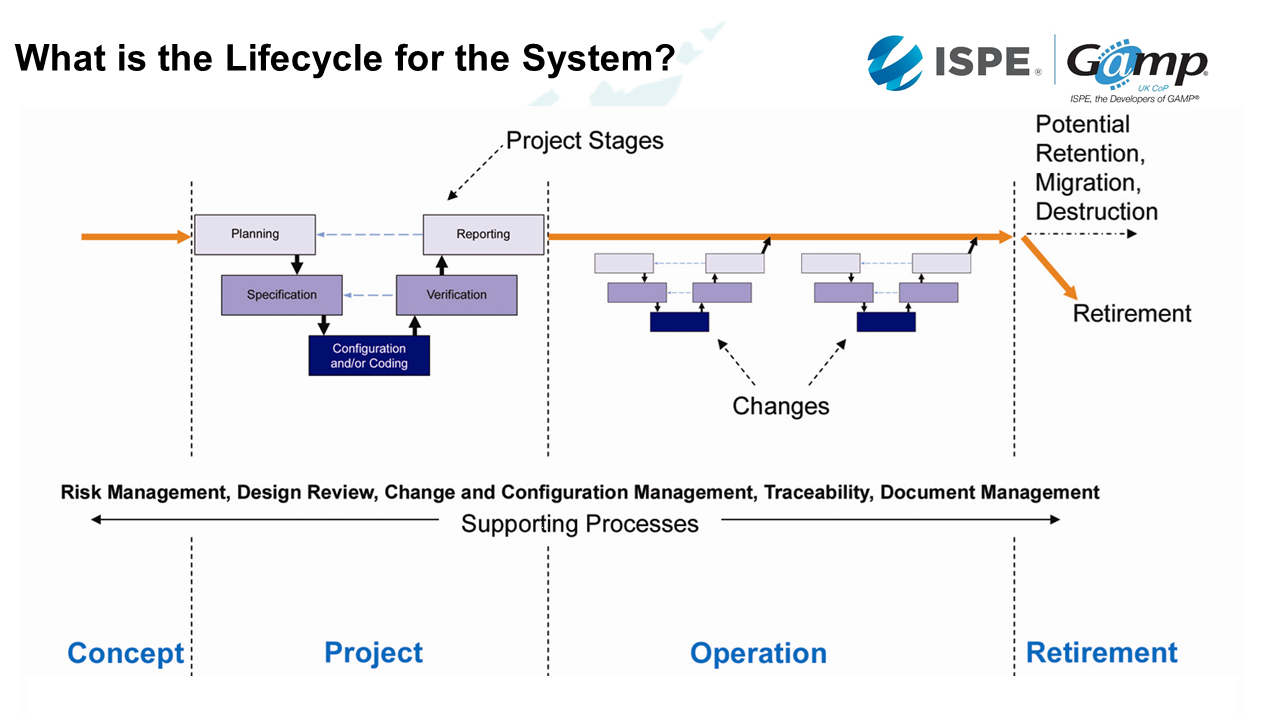
Concept phase
Concept is the phase where we start to think about it. This is usually while we're planning to develop a drug.
Project phase
Project phase is where we develop the computerised system. Here we start to plan, build our user requirements specification, and go through a design period. Then we start configuring or / and coding the tool. Once that's ready, we test and verify that it does what we need it to do. When we train on this, we always say it's very easy to write a test which doesn't actually test the specification - so make sure the tests are relevant!
Then, we need to check everything that we've done that it fills the requirements of the plan.
Operation phase
Lots of changes happen in the operation phase. We start to understand a bit more, there's more complexity and this means we need to update our computerised system. When these changes happen, we need to make sure the changes are controlled in the same way as they were in the project phase. We do this until we are ready to retire the system.
Supporting processes
Throughout the life cycle of the system, we need to have robust supporting processes, such as risk management, traceability, design review, change and configuration management, traceability, and document control.
Many companies use EQMS by Qualsys to holistically manage and embed supporting processes.
What is the life cycle for the data?
It's not just the systems which have a life cycle, data has a life cycle too.
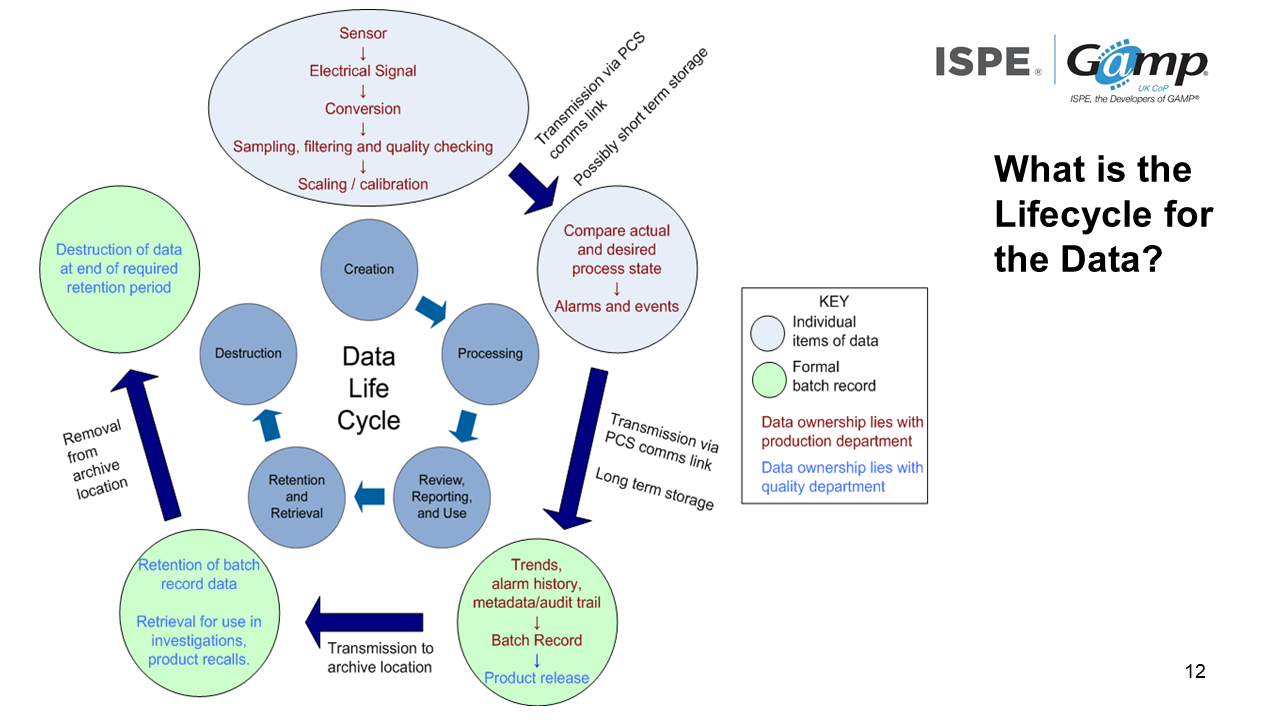
Data is almost certainly going to be processed in some way so that we can review, report and use it. Then we have a period in which the data can be used. Finally we can destruct it.
In terms of a manufacturing system, there are probably sensors, electrical signals etc. in order to get us the values that we are interested in. That's almost certainly being transmitted from your communications link. The data will then be processed so that we can understand it.
Then the data is almost certainly transmitted externally or internally once again. We can then look at trends and make up the batch records.
We can then release the product. After we've released the product, we need to be able to retrieve data. At the end of the retention period, we can then destruct the data.

Risks associate with functionality
We've got risks associated with the critical functions.
Patient safety:
We need to know whether there is anything in the drug which could harm somebody. For example, when they made the Polio vaccine in the 50's, they hadn't killed all the live virus and they injected people. The manufacturing process was to blame and it caused a product which went out which was unsafe to people. We need to plan for what we can do in the manufacturing process to prevent product risks.
Product quality:
We want the drug to do what it says on the tin. I have a friend who had a heart transplant in 1989. She has taken anti-rejection drugs since. If the drug product quality were to be low, and she rejects that vital organ, that's incredibly serious. We need to ensure that vital product quality.

Control the risks associated with the functionality
We need to plan how we will control the risks associated with the functionality of our manufacturing system. This will involve understanding the CQA (Critical quality attributes), CPP (Critical process parameters) and then ensuring this is covered in your URS.
For example, if we know the temperature is too low, we know there will be residual material that doesn't get converted into the product. The risk to the patient could cause mild nausea if it makes it's way into the final product - we might consider this a low risk. We may also find if it's at the right temperature but not for long enough, we won't convert all the material, resulting in the same issue. If the CPP temperature is too high, this is a high risk.
After we've made this assessment, we need to think about how we're going to manage these risks in a computerised system. With this scenario, we know what the risks are associated with it, for example, what if the temperature sensor fails? Can the operator change the temperature? In the risk assessment process, we need to control these issues, for example we can control the temperature manually, an alarm if there is high temperature, lock the setpoint entry and provide training and effective documentation.

Risks associated with the data
Our primary focus is what is the potential harm to the patient. What happens if we release our product that we think is good but our data is wrong?
What happens if we haven't got the right data to do a product recall when we need to?
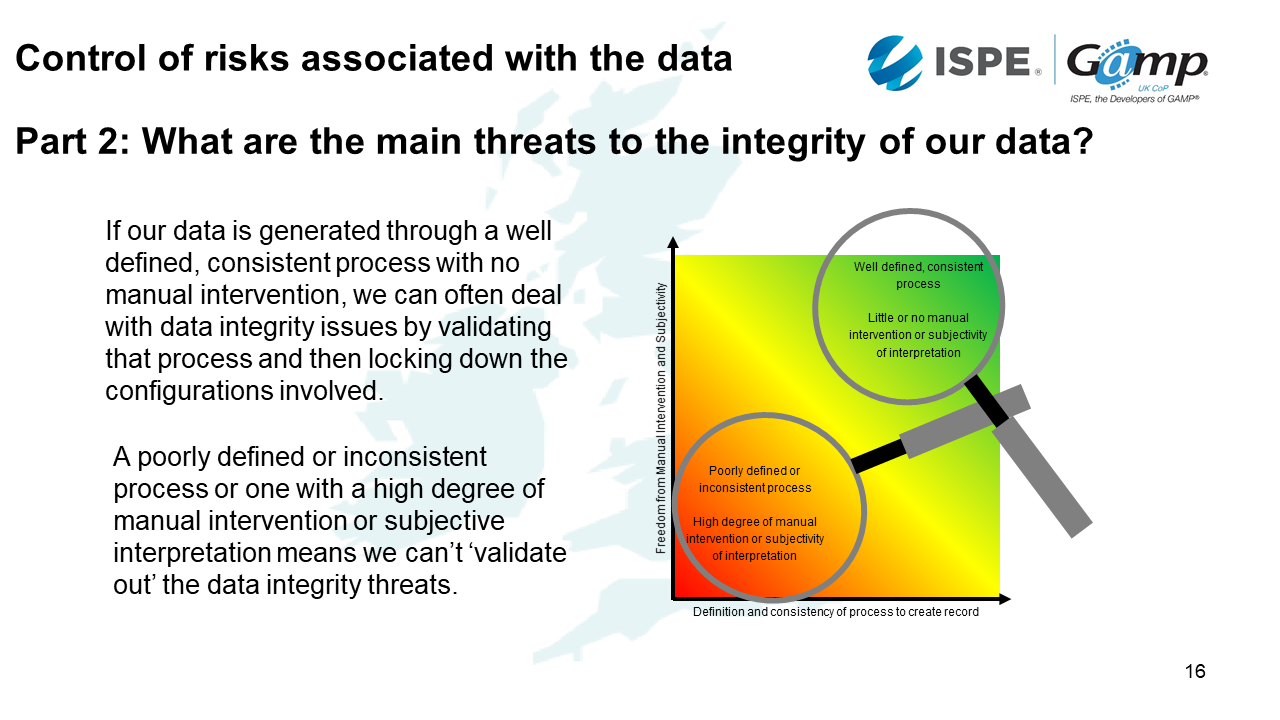
The second area we need to look at are the threats to data integrity.
This diagram is from the data integrity guidance. It shows how well-defined and consistent a process is versus what degree of manual intervention is involved.
For example, if we've got a well-defined, consistent process with no manual intervention and there is no difficulty in interpreting the data - this is probably not going to be a big threat to our data integrity because we can validate the system and lock down the configurations. On the other hand, if we've got a poorly defined or inconsistent process with a high degree of manual intervention or subjective interpretation, we can't validate out the data integrity threats.
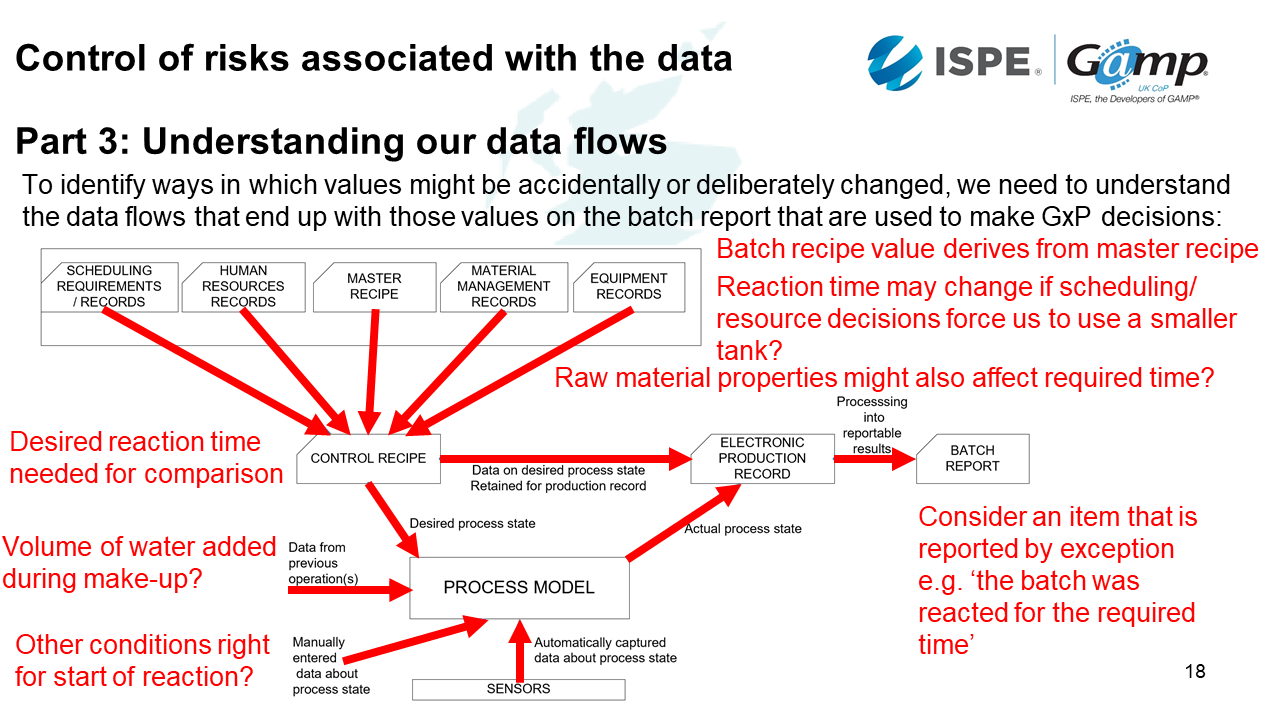
Control of risks associated with the data
To understand ways in which data values may be accidentally or deliberately changes, we need to understand how the data flows through the system or systems.
Scenario example: An item that is reported by exception e.g. The batch reacted for the required time
Imagine a temperature dial - we have some sensor information which is being passed through as an electronic production record. We may also have some manually entered data and data from previous operations. We will also need to know what the control recipe is and its desired range. That recipe is derived from a master recipe. We may also have inputs from scheduling, resourcing, and equipment. This may have an indirect impact on the batch because if the reaction time changes, and forces us to use a smaller tank. Finally, if raw material properties vary, this may also affect the required time.
The data can be wrong at any stage in the process. There might be noise which impacts the signal. This could be overcome through EMC standards, installation standards, filtering and validation.
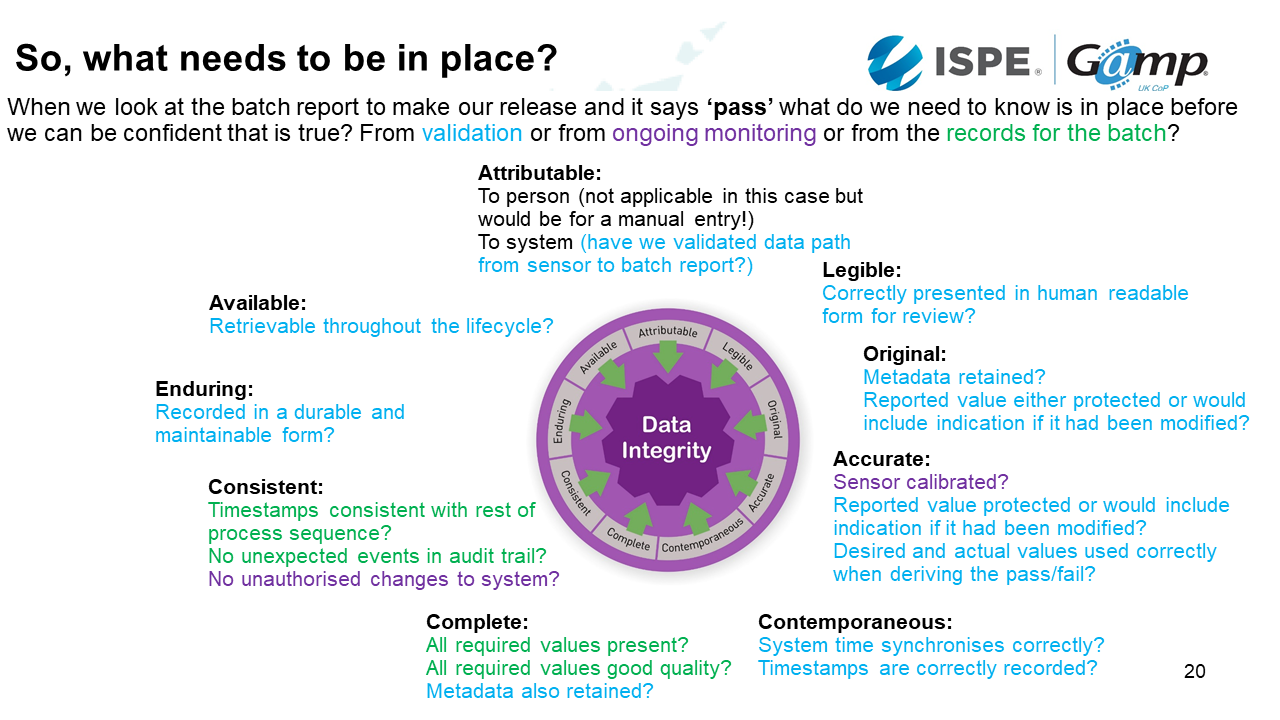
So, what needs to be in place?
To summarise, we've focused in this presentation on manufacturing systems used for automating drug production. Established methods from GAMP 5 for developing manufacturing systems with an understanding of CQA / CPP and the use of risk assessment. And we've looked at the trend for controlling data integrity for critical records using data flows and identifying threats.
|
Want all the resources from the GxP Life Science event? Complete this form:
|




Share your thoughts on this article